
什么是无菌
无菌是指在产品上不存在任何存活的微生物的状态。灭菌就是杀灭细菌及其细菌繁殖体、芽孢、病毒和真菌孢子等一切形式的微生物的过程。
大量研究证明,物理和化学因子对于微生物的杀灭遵循指数法则。随着灭菌作用时间的延长,产品上的微生物数量级近似于线性的下降。但是,任何产品经过任何灭菌过程,其携带的微生物始终有一定的存活概率。可以这样说,没有一种灭菌方法可以**保证无菌,而只能将存活概率降低到极低水平。《ISO 11135 医疗器械环氧乙烷灭菌的确认和常规控制》规定了无菌医疗器械的带菌概率需在10 -6 cfu以下,该概率被称为为无菌保证水平(Sterile Assurance Level ,SAL)。
环氧乙烷灭菌
环氧乙烷作为一种广谱低温灭菌剂,目前在世界各G广泛的应用于医院和工业*域。据统计,有50%以上的生产厂商选用环氧乙烷对其医疗器械产品进行灭菌。
环氧乙烷又称作氧化乙烯,分子式为C 2 H 4 O,分子量为44.05,在常温常压下为气态,具有很强的化学活性和穿透性,可以穿过微孔,到达产品深层,从而大大提高了灭菌效果。
影响环氧乙烷灭菌效果的因素通常包括温度、相对湿度、环氧乙烷浓度和时间。
在一定范围内,温度升高导致气体分子活动加剧,从而提高了环氧乙烷的灭菌效率。然而,在超过一定温度以后,灭菌效率的上升不明显。而且, 过高的温度也可能对产品造成损害。因此,通常选用的灭菌温度为40℃~60℃。
水是烷基化反应的反应剂,能打开环氧乙烷的环氧基团以使烷基化反应能顺利进行。另外,水能够加速环氧乙烷的穿透,提高灭菌效率。研究证明,湿度对于环氧乙烷灭菌成功起着非常关键的作用。比较理想的湿度范围是40%~80%,如湿度低于30%,容易导致灭菌失败。
在一定温度和湿度条件下,环氧乙烷的浓度升高可使灭菌效率提高。当然,过高的浓度并不能无限提高效率,反而会增加无谓的成本支出。当浓度大于500mg/L时,灭菌效率的提高已不明显。然而,考虑到环氧乙烷气体的损失,如水解和产品对气体的吸收,实际选择的浓度一般需高于理想作用浓度。
气体灭菌并非迅速的灭菌方式,而需经历一段时间才能达到灭菌效果。以上各参数的选择及产品、包装、装载形式等各因素的影响,都会导致所需灭菌时间的变化。
传统无菌检验的局限性
传统的监测方法为无菌检验,即抽取灭菌后的产品在无菌实验室中进行微生物培养,观察培养结果以确定该灭菌过程的有效性。然而,该种方式虽然能比较准确的反应被检测产品的带菌情况,但其在环氧乙烷灭菌过程监控方面的合理性和有效性存在以下的局限性。
无菌检验是一个抽检过程,其有效性建立在其抽样的合理性上。然而,由于环氧乙烷灭菌的影响因素很多、整个负载的均一性不高,无法找到可靠的抽样方案以准确反映整个负载的灭菌效果。
另外,无菌检验还有结果等待周期长、投入设备和人力大、报废产品成本高等缺点,从经济角度出发,也不是灭菌质量控制的理想方法。
生物监测
要寻找适合的灭菌监控手段,shou先需了解影响灭菌效果的主要因素。在灭菌环境相同的情况下,产品上负载的微生物的抗力和数量,是影响灭菌效率的关键因素。通常,由于对于原材料的来源和微生物的数量有严格要求,加上对于储存和生产车间的洁净要求(不同产品有不同要求),在灭菌前所携带的微生物的数量是有限的。生物监测使用所携带微生物的数量和抗力都远大于实际生物负载的生物指示剂,与生物负载处于相同或更难灭菌的环境中(如将生物指示剂放置于过程挑战设备{Process Challenge Device}中) 同时进行灭菌,然后观察生物指示剂中微生物的杀灭情况,来判断整个负载的有效性。图1解释了生物指示剂监控整个负载并可靠反映其效果的原理:当生物指示剂被杀灭的时候,生物负载早已被杀灭了。
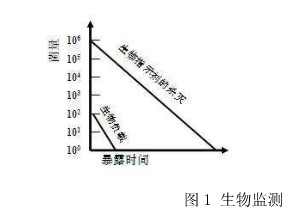
图1 生物监测
生物指示剂
当前用于灭菌效果验证和监测的生物指示剂,主要有菌片、自含式生物指示剂和芽孢悬浮液等几种形式 。
菌片因其价格便宜而应用广泛。在灭菌结束后,将菌片转移**配制好的培养基中进行培养,培养**少7天后观察微生物的生长情况,来判断灭菌效果。
自含式生物指示剂,是将菌片和培养基集成在一个塑料小瓶中,在灭菌结束后,从瓶外捏破装有培养基的玻璃安瓿,使培养液充分浸润内置的菌片,经培养后观察其变色情况或荧光反应来判断灭菌效果。
与菌片相比,自含式生物指示剂有以下优点:
**,菌片灭菌后培养过程繁琐,需经历配制培养基、培养基灭菌、无菌转移和培养等一系列过程;而自含式生物指示剂,含内置无菌培养液,操作简便,大大简化了操作人员的培训过程。
第二,菌片无菌转移过程中,如试样被污染,则培养结果存在假阳性的可能。而假阳性是很难判断的,因此一旦出现阳性,整批产品需重新进行灭菌,耗时耗力。而自含式生物指示剂采用密闭结构,其盖中的过滤材料能透过环氧乙烷气体和水蒸气,可有效的阻隔微生物的侵入,避免假阳性的出现。
第三,菌片的培养时间较长,**少需要7天培养时间。而自含式生物指示剂只需2天,即能反映灭菌效果,能够满足市场对于企业日渐增强的快速供给的要求。
第四,通过颜色变化来判断培养结果,相比通过混浊与否来观察更明确、更容易判断。
验证和常规监测
根据ISO11135规定,不能由随后的产品检验和试验来充分证实其结果的过程称为特殊过程。灭菌过程就是这种特殊过程,通过前面的论述可知,包括无菌检验在内的产品检验方法无法充分证明其结果——即无菌保证水平——的达到。对于这样的工艺过程,必须进行预先的验证和日常的监控。
灭菌验证,即是通过物理和生物实验的方法,证明所设定的灭菌工艺过程及参数,能够保证产品灭菌工艺要求的达到。ISO11135规定, 验证必须**少每年进行一次 。而且,一旦发生可能影响**终灭菌效果的改变——如更换包装、改变装载顺序等——发生时,必须进行验证。验证的过程通常牵涉企业众多部门,步骤繁多,耗时较长。简要的验证流程如下:
1) 验证前准备
需要制定验证方案,并由经过培训的人员来实施整个验证。对于产品灭菌的适用性、包装、灭菌剂、加湿蒸汽、生物指示剂等的适用性均需进行验证并形成文件。
2) 安装验证
需要验证的内容包括设备的相关资料和附件是否齐全、安装场所是否符合安全要求、对主要计量器具进行校验等。计量器具的校验必须shou先进行。
3) 运行验证(试运行)
设备安装后,应按操作说明书启动设备,确定设备是否能在预期的设计范围内准确的运行,并能达到各项技术指标和使用要求。
对于预处理区,需验证其空载状态下的温湿度均匀性;对于灭菌柜室,需验证其空载状态下的温度均匀性。
4) 物理性能验证
在满载状态下,验证设备达到预期工艺参数的能力。对于预处理区,需验证其满载状态下的温湿度均匀性;对于灭菌柜室,需验证其满载状态下的温度均匀性。
5) 微生物性能验证
可以认为,之前的验证目的在于确保设备主体及其他辅助用品能够满足工艺要求。而微生物性能验证的目的在于,在这样的条件下,寻找并证明能够满足灭菌工艺要求——10 -6 无菌保证水平——的工艺参数。
在选定的温度、相对湿度和环氧乙烷浓度的条件下,能将生物指示剂的存活概率降低到10 -6 或以下的气体作用时间,就是所需寻找的工艺参数。工业上,通常用半周期法来确定所需的灭菌时间。通过将灭菌时间依次减半,直**找到**短灭菌时间——即刚好能将生物指示剂杀灭的时间。用该时间重复两次灭菌过程,需全部阴性。由于根据《ISO11138 医疗保健产品灭菌生物指示物》的规定,用于环氧乙烷灭菌验证和监测用的生物指示剂的**小菌量需达到1.0×10 6 ,该**短灭菌时间的两倍,即为能满足灭菌要求的气体作用时间,如图2所示。
需要提醒的是,半周期法虽然是**简便的方法,却并不是**可靠的。由于严格来讲,对于生物指示剂的杀灭曲线是未知的,半周期法的结果只是建立在推测的基础上。所以,有条件的企业,应该选用更严格、更可靠的存活曲线法或部分阴性法。
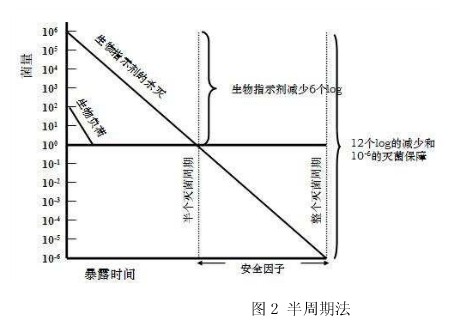
图2 半周期法
验证时生物指示剂需摆放在整个负载**难灭菌的部位,并应达到一定的数量以全面反应灭菌柜室内各位置的灭菌效果。对于生物指示剂的摆放数量,ISO11135有如下的推荐:
☆ 柜室体积达到5m3 时,**少20支
☆ 柜室体积5 m3 -10m3 时,每增加1m3 ,增加2支
☆ 柜室体积>10m3 时,每增加2m3 ,增加2支
常规监测用于对验证后的日常灭菌过程进行监测,控制产品放行。建议使用的生物指示剂的数量通常为验证时的一半。
当前存在的问题
目前在G内,由于经费的限制及认识的不足,对于工业上灭菌流程的规范,尚存在以下一些问题。
1) 验证的不科学
“一招鲜”的验证方法,在G内还十分常见。所谓“一招鲜”,就是只对一种产品、一台设备、一个装载方式进行验证,然后将这套灭菌工艺用于所有的产品、设备和装载方式。
这主要是由于对验证的认识不足导致的。验证得到的灭菌参数只能用于该次验证的灭菌工艺,因为不同的产品、设备、装载等因素,都会对灭菌效果产生影响。对于不同的灭菌过程,没有任何一套灭菌参数是可以确保灭菌效果的。必须对任何不同的灭菌过程分别的进行验证,确定有科学依据的、可确保灭菌效果的灭菌参数。
另外,有很多企业未严格遵守ISO标准规定的验证流程,文件系统的不规范、对于设备的验证不充分等问题仍然存在。
2) 常规监测的不重视
对于经过验证的灭菌过程,很多企业在日常的灭菌流程上,选择了参数放行、化学监测或无菌检验的方法来放行产品。以上这些控制方式,对于工业环氧乙烷灭菌来说,都是不充分的。
影响环氧乙烷灭菌效果的因素很多且相互关联,控制起来十分困难。
例如,湿度就是一个多变因素。即使经过了有效的预处理,由于环境湿度的不稳定、抽真空湿度损失等原因,灭菌作用过程中分散到每个包裹的湿度仍是不可预知和不可控制的。如使用混合环氧乙烷气体,由于气体的均匀性的不稳定,实际作用在产品上的环氧乙烷浓度也是很难精确计算和控制的。另外,还有很多未知因素影响着环氧乙烷灭菌的有效性。因此,即使通过验证确定了参数,仍旧要对日常的灭菌进行常规监测,确认灭菌实际有效后才能放行。
化学指示剂是通过灭菌过程对于化学物品的作用导致的化学变化,来模拟生物指示剂的灭活。然而,化学指示剂并不能代替生物指示剂的作用,**终能确定整个负载灭菌效果只有生物监测。
3) 生物指示剂的选择和用法不当
用作灭菌验证和常规监测的生物指示剂,需谨慎的选择。性能符合ISO11138的规定并获得上市许可,是选择生物指示剂的基本条件。需要注意的是,如无菌产品将**终出口,所选择的生物指示剂也需满足该G的要求并获得认可。
对于生物指示剂的使用,目前也存在着用量不足和用法不当等问题。
在灭菌柜室中使用足够多的生物指示剂进行全面的监测是必须的。然而,出于成本的考虑,一些企业未使用足够多的生物指示剂进行验证和常规监测。其实,这样所得的结果是不够可靠的。因为无论是在验证还是常规监测时,只有全面监控整个灭菌柜室的情况,才能使所得灭菌参数真正能够保证灭菌的有效性。
另有一些企业,在验证和常规监测时使用不同的生物指示剂或混用不同品牌的生物指示剂。由于不同的生物指示剂的菌量和抗力有所不同,这对于验证和常规监测的一致性是有影响的。
将生物指示剂放在**难灭菌部位,也是**关重要的。PCD为灭菌过程对生物指示剂的杀灭提供**大挑战,是保证生物监测的有效性的重要手段。对PCD的可靠性,亦需进行验证。
总结
由于环氧乙烷灭菌的过程复杂、导致其失败的原因很多,并且没有经典的参数配置可以适用于各种情况,为了保证灭菌的质量,对其进行严格的验证和常规监测是必要的。
生物监测覆盖了整个负荷,并能确实保证SAL的达到,是**能够全面确保 灭菌效果 的监测方式。
在高速发展的中G社会,对医疗服务与产品的质量诉求不断增高。对于日渐成熟的中G医疗产品企业,更多的与G际接轨、与各G企业开展贸易的趋势不可阻挡。无菌医疗产品的灭菌效果控制,是其质量控制中的重中之重。不使用科学的、规范的手段确保灭菌效果的医疗产品,既很难满足G内市场日益增高的质量诉求,也无法在G际市场上获得认可。因此,规范灭菌工艺、确保灭菌质量,成为无菌医疗产品生产企业的当务之急。


